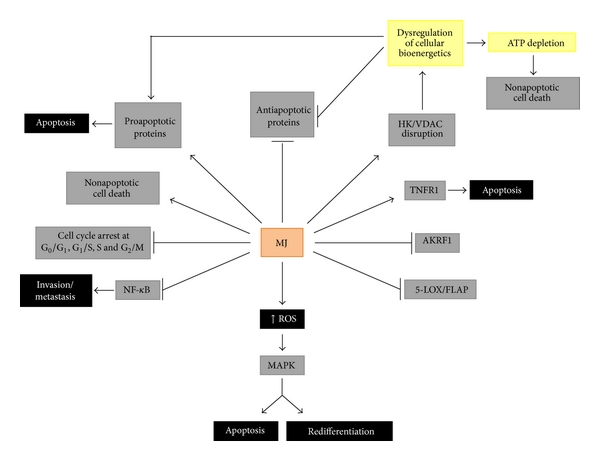
Update: Follow the link for a nice primer on apoptosis, BCL-2, and BH3 Mimetics.
Dr. Sharman’s CLL & Lymphoma Blog – What is BCL-2 and why should we inhibit it?
“Chronic lymphocytic leukemia (CLL) is characterized by the deregulated accumulation and persistence of B lymphocytes in the blood. Although the exact causes of CLL are unknown, the evasion of apoptosis through aberrant expression of BCL2-family proteins is a common feature. A class of compounds, termed BH3 mimetics, has been developed to directly inhibit BCL2 proteins and selectively kill tumor cells. To date, the most successful of these compounds are the BCL2/BCLXL inhibitors ABT-7372 and ABT-263 (navitoclax), as well as the BCL2-specific inhibitor ABT-199. Results from early clinical trials with navitoclax have demonstrated single-agent efficacy in patients with relapsed or refractory CLL.
However, there was heterogeneity in response rates between patients, and dose-limiting toxicities including thrombocytopenia and neutropenia which prevented further doseescalation.
In addition, CLL cells residing within various microenvironments (e.g. lymph nodes and bone marrow) are resistant to BCL2 inhibitors. This resistance results from the upregulation of additional BCL2-proteins, such as BCLXL, MCL1 and BFL1, the latter two of which are not inhibited by navitoclax, and therefore protect the leukemia cells from apoptosis. Additional drugs are needed to enhance the efficacy of navitoclax. Here, we demonstrate that gossypol overcomes stroma-mediated resistance to ABT-737 without enhancing the sensitivity of normal lymphocytes and platelets.
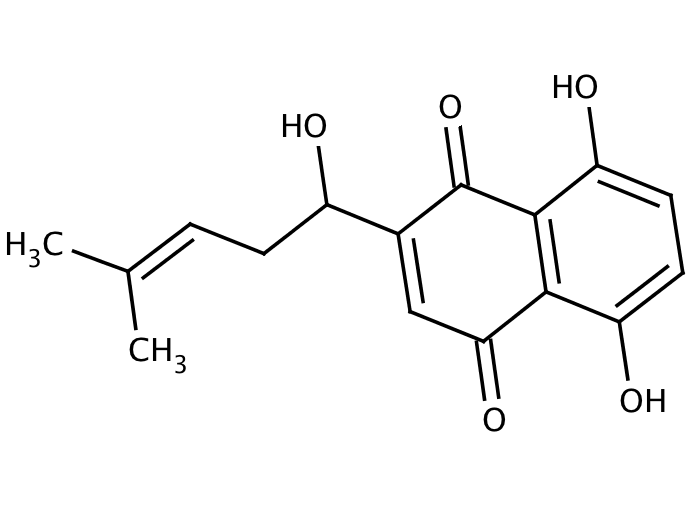
The BH3-only protein, NOXA, is a potent inhibitor of MCL1 and BFL1, but has recently been recognized to inhibit BCLXL with lower affinity. Therefore, compounds which induce NOXA may inhibit MCL1, BFL1 and BCLXL, thus overcoming resistance to navitoclax. We previously reported that six putative BH3 mimetics do not directly inhibit BCL2 in cells, but instead activate the integrated stress response and induce NOXA. Of these six compounds, gossypol has advanced into clinical trials in a racemically purified form (AT-101).10 We hypothesized that gossypol, through induction of NOXA, would sensitize CLL cells to ABT-737”. Link below:
Gossypol overcomes stroma-mediated resistance to the BCL2 inhibitor ABT-737 in chronic lymphocytic leukemia cells ex vivo
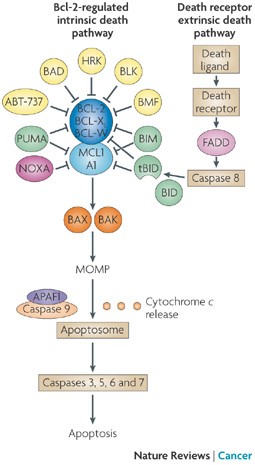
BH3 Mimetics – the road to apotosis
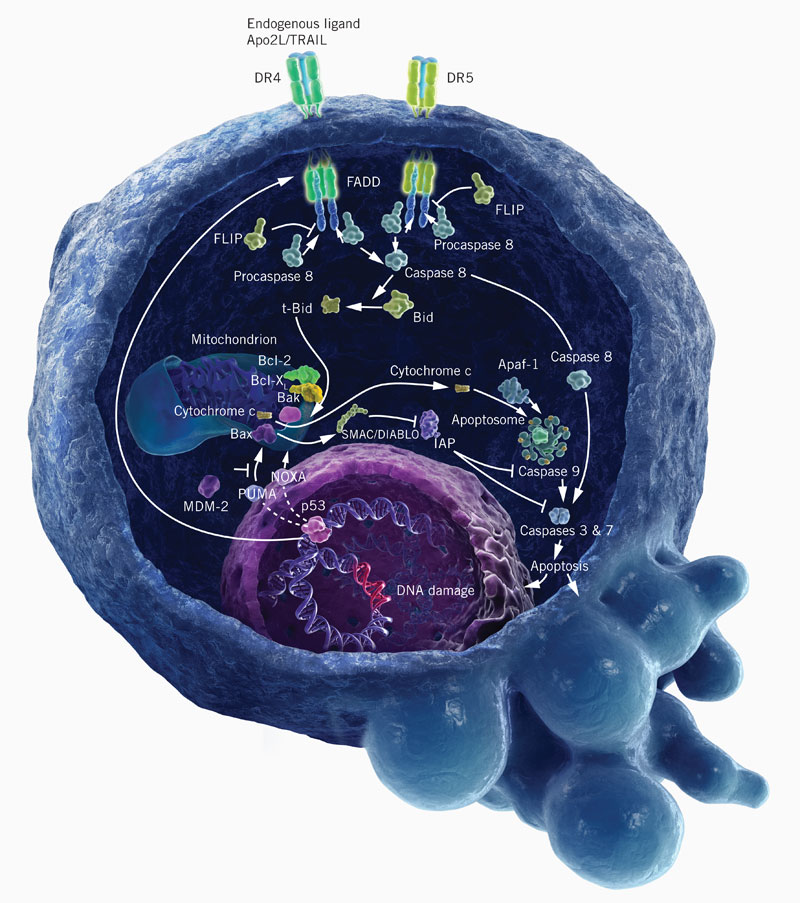
“In mammals, apoptosis occurs through the death receptor (extrinsic) or Bcl-2-regulated (intrinsic or mitochondrial) pathways. The latter is regulated by three subgroups of the Bcl-2 family: the pro-survival members, such as BCL-2 or MCL1, the pro-apoptotic BAX and Bcl-2 homologous killer (BAK) subgroup and the pro-apoptotic BCL-2 homology domain 3 (BH3)-only proteins, such as BIM (also known as BCL2L11) and PUMA (also known as BBC3). Apoptotic stimuli cause transcriptional and/or post-translational activation of specific BH3-only proteins, which then engage and sequester the pro-survival Bcl-2 family members, thereby liberating the downstream effectors, BAX and BAK, which elicit mitochondrial outer membrane permeabilization (MOMP) and unleash the caspase cascade, culminating in cell demolition. It has also been proposed that at least some BH3-only proteins, in particular BIM and BID, can directly activate BAX and BAK (not shown). Some BH3-only proteins (shown in green), such as BIM and PUMA, can bind and sequester all anti-apoptotic Bcl-2 family members with high affinity and are thus potent killers, whereas others (shown in yellow and dark pink), such as Bcl-2 antagonist of cell death (BAD) and NOXA (also known as PMAIP1), bind only certain anti-apoptotic members (BAD binds BCL-2, BCL-XL and BCL-W (dark blue), whereas NOXA binds only MCL1 and A1 (light blue)). Thus, the efficiency of cell killing is determined by the relative levels of pro- and anti-apoptotic members. ABT-737, a BH3-mimetic, has a similar binding profile to the BH3-only protein BAD. APAF1, apoptotic protease-activating factor 1; BMF, Bcl-2-modifying factor; HRK, activator of apoptosis harakiri; tBID, truncated BID.”
Links:
Multiple BH3 Mimetics Antagonize Antiapoptotic MCL1 Protein by Inducing the Endoplasmic Reticulum Stress Response and Up-regulating BH3-only Protein NOXA
Development of Noxa-like BH3 Mimetics for Apoptosis-Based Therapeutic Strategy in Chronic Lymphocytic Leukemia
Apoptosis: from biology to therapeutic targeting
Gossypol, a BH3 mimetic, induces apoptosis in chronic lymphocytic leukemia cells